Direct effect sizes estimated from latent factor models
This function returns 'direct' effect sizes for the regression of X (of dimension 1) on the matrix Y, as usually computed in genome-wide association studies.
effect_size(Y, X, lfmm.object)
Arguments
| Y | a response variable matrix with n rows and p columns. Each column is a response variable (numeric). |
|---|---|
| X | an explanatory variable with n rows and d = 1 column (numeric). |
| lfmm.object | an object of class |
Value
a vector of length p containing all effect sizes for the regression of X on the matrix Y
Details
The response variable matrix Y and the explanatory variable are centered.
Examples
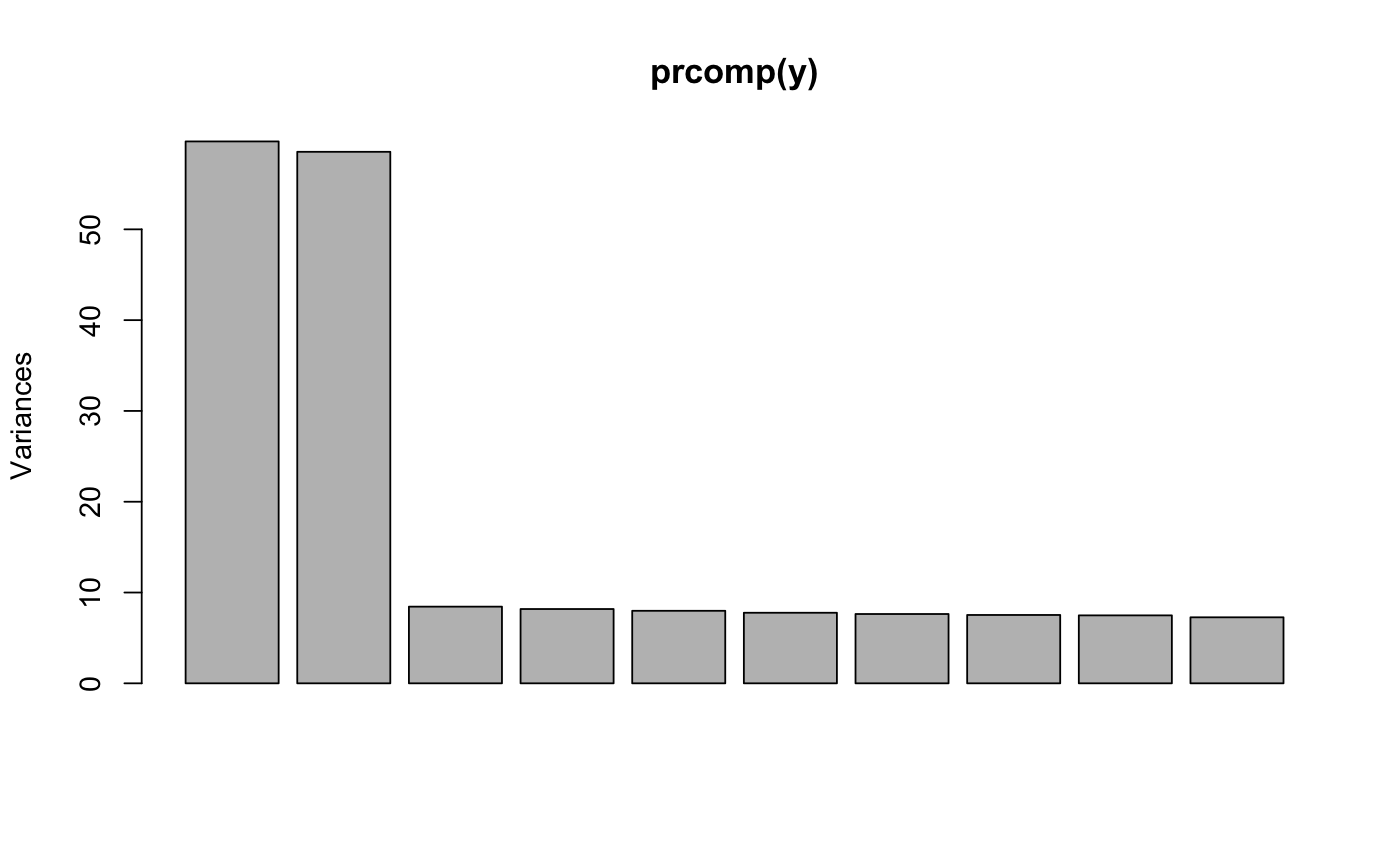
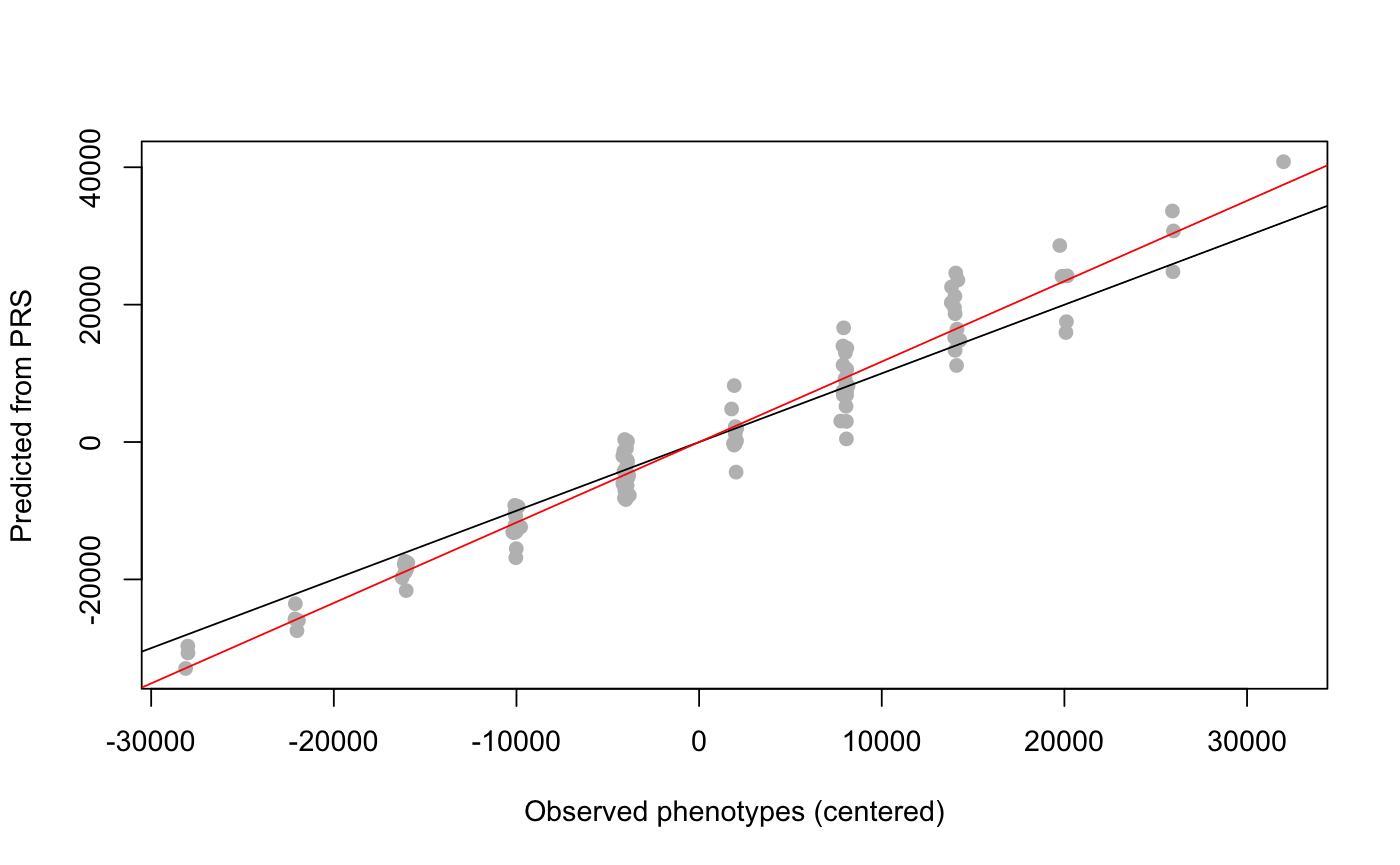
library(lfmm) ## Simulation of 1000 genotypes for 100 individuals (y) u <- matrix(rnorm(300, sd = 1), nrow = 100, ncol = 2) v <- matrix(rnorm(3000, sd = 2), nrow = 2, ncol = 1000) y <- matrix(rbinom(100000, size = 2, prob = 1/(1 + exp(-0.3*(u%*%v + rnorm(100000, sd = 2))))), nrow = 100, ncol = 1000) #PCA of genotypes, 3 main axes of variation (K = 2) plot(prcomp(y))## Simulation of 1000 phenotypes (x) ## Only the last 10 genotypes have significant effect sizes (b) b <- matrix(c(rep(0, 990), rep(6000, 10))) x <- y%*%b + rnorm(100, sd = 100) ## Compute effect sizes using lfmm_ridge ## Note that centering is important (scale = F). mod.lfmm <- lfmm_ridge(Y = y, X = x, K = 2) ## Compute direct effect sizes using lfmm_ridge estimates b.estimates <- effect_size(y, x, mod.lfmm) ## plot the last 30 effect sizes (true values are 0 and 6000) plot(b.estimates[971:1000])abline(0, 0)abline(6000, 0, col = 2)## Prediction of phenotypes candidates <- 991:1000 #set of causal loci x.pred <- scale(y[,candidates], scale = F) %*% matrix(b.estimates[candidates]) ## Check predictions plot(x - mean(x), x.pred, pch = 19, col = "grey", xlab = "Observed phenotypes (centered)", ylab = "Predicted from PRS")abline(0,1)abline(lm(x.pred ~ scale(x, scale = FALSE)), col = 2)