Estimating cell-type composition accounting for confounder variables.
Michael Blum, Clémentine Decamps, Florian Privé, Magali Richard
June 6, 2019
deconvolution.RmdBased on DNA methylation values, the vignette shows how to estimate cell-type composition and cell type-specific methylation profiles accounting for confounder variables such as age and sex.
Data
We provide access to a simulated matrix D of a complex tissue DNA methylation values. The data frame exp_grp contains the corresponding experimental data for each patient. Here the data frame contains 22 different variables that include sex and age.
## [1] 23381 20head(D)[1:5, 1:5]## [,1] [,2] [,3] [,4] [,5]
## cg00000292 0.70429380 1.0000000 0.97145801 1.0000000 0.83341004
## cg00002426 0.09539247 0.2213554 0.06816716 0.0233084 0.09012396
## cg00003994 0.06072960 0.3590995 0.01321180 0.2115948 0.18793080
## cg00005847 0.71071274 1.0000000 1.00000000 1.0000000 1.00000000
## cg00007981 0.00611625 0.0792212 0.02016318 0.3125305 0.01918717## [1] 20 22head(exp_grp)[1:5, 1:5]## organism sample_source cigarette_exposure tissue_stage t
## 1 Homo sapien Primary Tumor NA adult T1a
## 2 Homo sapien Primary Tumor NA adult T2
## 3 Homo sapien Primary Tumor 60 adult T1
## 4 Homo sapien Primary Tumor 20 adult T1b
## 5 Homo sapien Solid Tissue Normal 34 adult T2aStep 2: choosing the number of cell types K
To choose the number \(K\) of cell types, we used the function plot_k, which performs a Principal Component Analysis and plot the eigenvalues of the probes matrix in descending order.
medepir::plot_k(D_CF) 
To select the number of principal components, we use Cattell rule that recommends to keep principal components that correspond to eigenvalues to the left of the straight line. Here, Cattell rule suggests to keep 4 principal components which corresponds to 5 cell types (the number of cell types is equal to the number of principal components plus one).
Step 3: feature selection
This step is optional. The function feature_selection select probes with the largest variance (5000 probes by default). By removing probes that do not vary, deconvolution routines can run much faster.
D_FS = medepir::feature_selection(D_CF)
dim(D_FS)## [1] 4999 20Step 4: running deconvolution methods
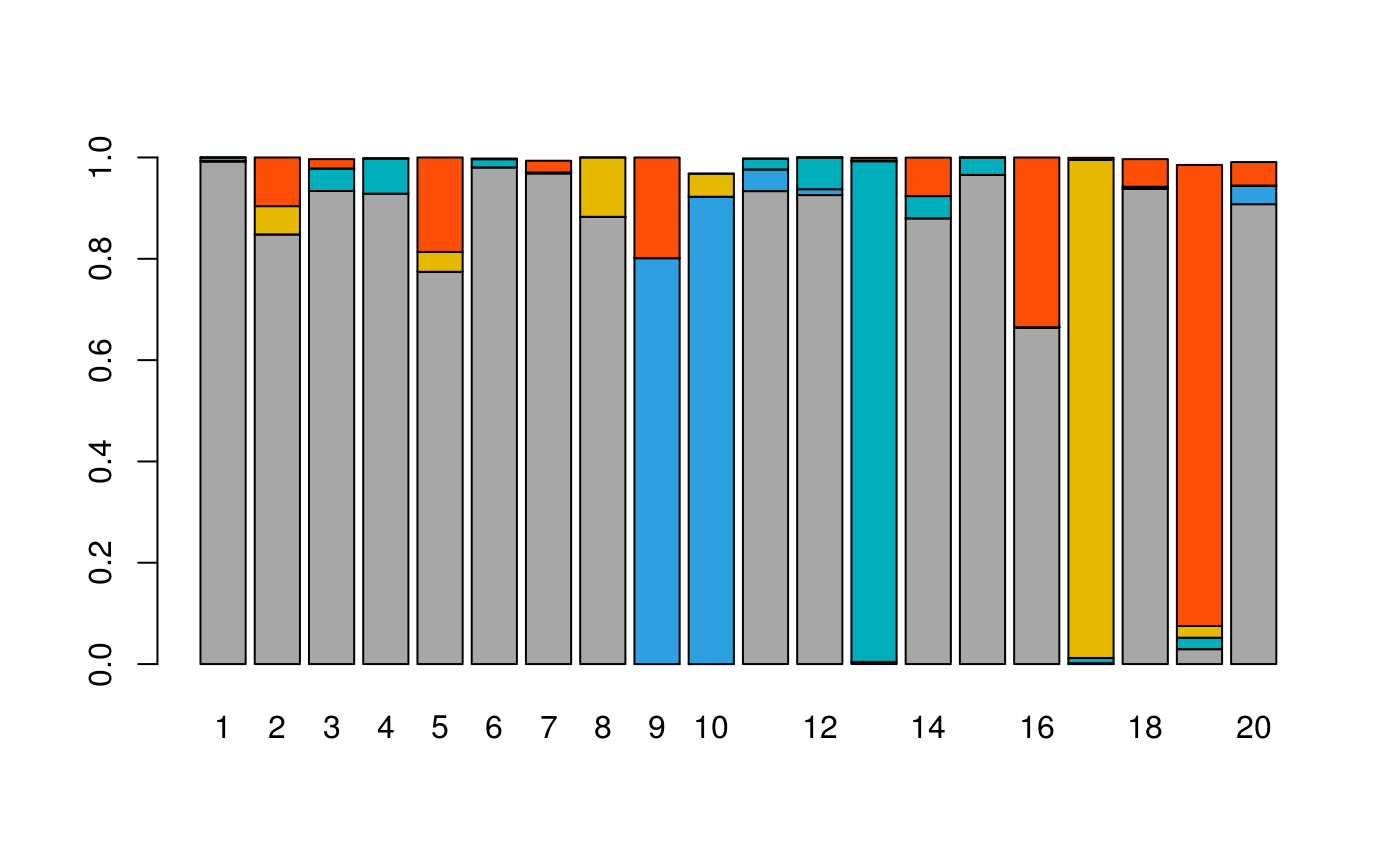
We propose three methods of deconvolution, of three packages: RFE to run RefFreeEWAS::RefFreeCellMix, MDC to run MeDeCom::runMeDeCom and Edec to run EDec::run_edec_stage_1.
Here we show the results obtained with RefFreeEWAS.
results_RFE = medepir::RFE(D_FS, nbcell = 5)## The "ward" method has been renamed to "ward.D"; note new "ward.D2"## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 4.600e-07 2.266e-03 5.265e-03 8.524e-03 1.110e-02 8.599e-02
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.000000 0.001734 0.003882 0.006065 0.007735 0.061033
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.000000 0.001343 0.003002 0.004482 0.005753 0.043014
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.000000 0.001088 0.002383 0.003300 0.004391 0.028942
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.000000 0.000873 0.001899 0.002490 0.003440 0.019102
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.0000000 0.0007163 0.0015768 0.0020071 0.0028159 0.0136193
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.0000000 0.0006118 0.0013481 0.0016938 0.0023925 0.0106228
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.0000000 0.0005413 0.0011860 0.0014716 0.0020968 0.0083315
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.0000000 0.0004882 0.0010600 0.0013014 0.0018606 0.0069595Results obtained with EDec and MeDeCom can be obtained by uncommenting the following code.
#results_MDC = medepir::MDC(D_FS, nbcell = 5, lambdas = c(0, 10^(-5:-1)))
#infloci = read.table(url("https://zenodo.org/record/3247635/files/inf_loci.txt"),header = FALSE, sep = "\t")
#infloci = as.vector(infloci$V1)
#results_Edec = medepir::Edec(D, nbcell = 5, infloci = infloci)