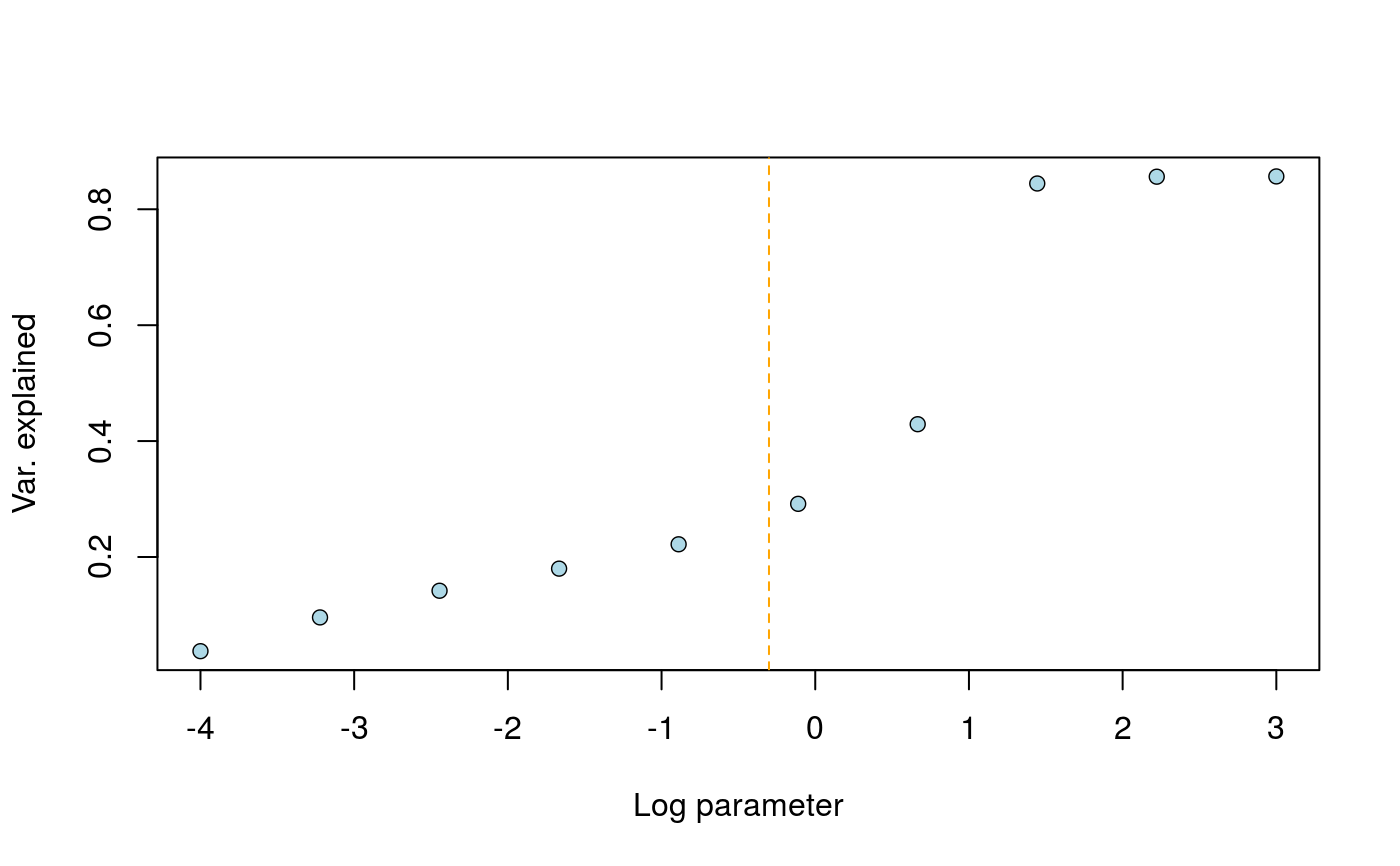
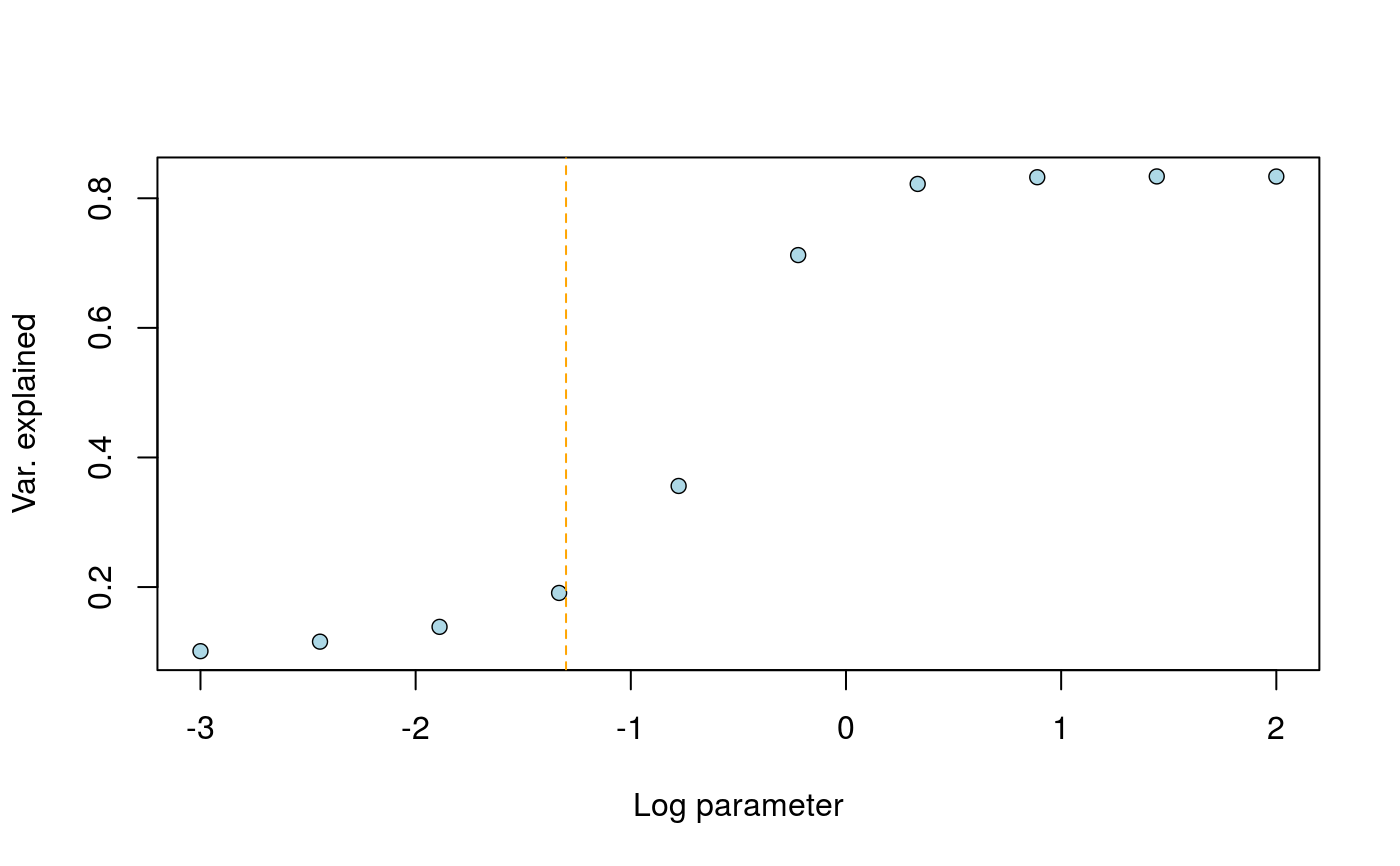
Graphical heuristic for choosing the drift parameter in tfa()
choose_lambda.Rdchoose_lambda runs temporal factor analyses for drift parameters in a specified
range, and ouputs the percentage of variance of sample time explained by all factors for each value.
choose_lambda(model, Y, min_range = -5, max_range = 1, grid_size = 10, plot_res = TRUE, detail_axis = FALSE)
Arguments
| model | an object of class 'tfa' adjusted with |
|---|---|
| Y | an nxp numeric matrix containing genetic information for n individuals recorded in p columns. Genetic information could be encoded by any numeric value, not necessarily an integer value. Missing data are not allowed. |
| min_range | a numeric value for the minimal range of drift parameter tested. Log 10 scale.
The default value is |
| max_range | a numeric value for the maximal range of drift parameter tested. Log 10 scale.
The default value is |
| grid_size | an integer value for the number of drift parameter tested. The default is
|
| plot_res | a logical indicating whether the results should be displayed graphically or not. The
default value is |
| detail_axis | a logical indicating whether the results should also be
displayed for each factor independently. The default value is |
Value
A vector of percentages of variance of sample time explained by factors for each value of the drift parameter in the specified range.
Details
This function requires that a preliminary model has been adjusted with K factors, where K is the number of source populations. The curve exhibits a drop for some value in the range, and the heuristic consists of choosing the largest values corresponding to removing time variation in factor K.
References
François, O., Liégeois, S., Demaille, B., Jay, F. (2019). Inference of population genetic structure from temporal samples of DNA. bioRxiv, 801324. https://www.biorxiv.org/content/10.1101/801324v3
François, O., Jay, F. (2020). Factor analysis of ancient population genomic samples. Under review.
See also
Examples
library(tfa) # Ancient DNA from Bronze Age Great Britain samples data(england_ba) attach(England_BA) coverage <- meta$Coverage geno <- coverage_adjust(genotype, coverage, K = 4, log = TRUE) # Adjust an FA model mod <- tfa(age, geno, k = 3, lambda = 5e-1, center = TRUE, coverage = coverage, log = TRUE) r_2 <- choose_lambda(mod, geno, min_range = -4, max_range = 3)# Remove HG from Serbia to keep k = 2 ancestral populations age2 <- age[meta$Group.ID != "Serbia_HG"] geno2 <- geno[meta$Group.ID != "Serbia_HG",] # Adjust an FA model mod <- tfa(age2, geno2, k = 2, lambda = 5e-2, center = TRUE) r_2 <- choose_lambda(mod, geno2, min_range = -3, max_range = 2)